دانلود مقاله ساختار میتوکندری در انسان و نقش آن در ایجاد بیماریهای مختلف میتوکندری
مقدمه:
اولین گزارشات در ارتباط با ساختارهای درون سلولی شبه میتوکندری به ۱۵۰ سال پیش برمیگردد. واژه میتوکندری که از دو کلمه یونانی mitos بمعنی نخ یا رشته و chondros به معنی گرانول منشا گرفته است؛ برای اولین بار صد سال پیش مورد استفاده قرار گرفت. عملکرد اصلی این ارگانل کروی یا میلهای شکل که صدها عدد از آن در یک سلول وجود دارد، فسفریلاسیون اکسیداتیو است؛ بعبارت دیگر اکسیداسیون سوبستراها به Co2 و آب و فراهم کردن ترکیب پرانرژی ATP برای سلولها؛ و به همین دلیل است که میتوکندری را نیروگاه یا موتورخانه سلول نیز مینامند. بیماریهای دژنراتیو بسیار زیادی تا به امروز با نارساییها و اختلالات میتوکندری مرتبط شدهاند. این بیماریها میتوانند در اثر موتاسیون در DNA میتوکندری و یا DNA هسته ایجاد شوند. اولین بیماریهای میتوکندریایی که در سطح ملکولی درک شدند؛ در یک بیمار CPEO (فلج مزمن پیشرونده عضلات چشمی خارجی) و KSS (سندرمkearns-sayre) گزارش شدند. در همان زمان wallace موتاسیونی نقطهای را در ژن ND6 گزارش کرد که با LHON (نوروپاتی چشمی ارثی لبر) مرتبط است. در سال ۱۹۹۰، دوموتاسیون جدید، یکی در ژن لایزیل- tRNA در سندرم MERRF و دیگری در ژن لوسیل – tRNA در سندرم MELAS گزارش شدند. طیف فتوتیپی بیماریهای میتوکندریایی از میوپاتیهای نادر تا بیماریهای متعدد را شامل میشود. برخی موتاسیونهای mtDNA، علائم و نشانههای منحصر و ویژهای دارند؛ مثل جهشهای اشتباهی که موجب نوروپاتی چشمی ارثی لبر میشوند در حالیکه بقیه تظاهرات مولتی سیستم متنوعی را شامل میشوند مثل جهشهای حذفی که موجب CPEO میشوند. بیماریهای میتوکندریایی بواسطه وراثت مادری، وراثت منرلی و نیز نوترکیبیهای دوتایی نو، قادر به انتقال میباشند. این پیچیدگی ژنتیکی از این حقیقت ناشی میشود که میتوکندری از حدود ۱۰۰۰ ژن که در بین ژنوم میتوکندری و هسته پخش شدهاند، تشکیل شده است. علاوه بر این بیماریهای میتوکندریایی غالباً شروع تاخیری و یک دوره پیش رونده دارند که احتمالاً از تجمع جهشهای سوماتیک mtDNA در بافتهای post-mitotic حاصل شدهاند. این موتاسیونهای سوماتیک mtDNA همچنین در سرطان و پیری نیز نقش دارند. اگرچه بیماریهای میتوکندریایی هر ارگانی را ممکن است درگیر کنند اما این بیماریها غالباً CNS، عضلات اسکلتی، قلب، کلیه و سیستمهای اندوکرین را تحت تاثیر قرار میدهند. علت این پیچیدگیهای فتوتیپی، نقش مهم میتوکندری در انواع پروسههای سلولی شامل تولید انرژی سلولی بوسیله فسفریلاسیون اکیداتیو، تولید گونههای سمی فعال اکسیژن (ROS) بعنوان یک محصول جانبی در فسفریلاسیون اکسیداتیو و تنظیم شروع آپوپتوزاز طریق فعال شدن نفوذپذیری پورهای انتقالی میتوکندری (mtPTP) است. (۱۹، ۲۰ و ۲۴)
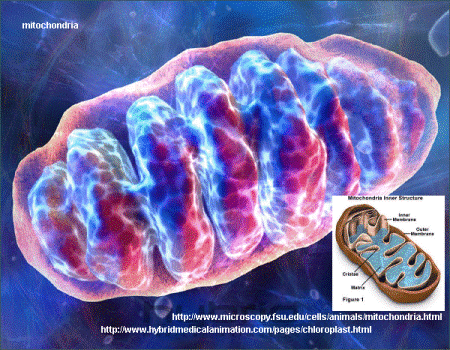
ساختار میتوکندری :
میتوکندری واجد یک غشای بیرونی و یک غشای داخلی است که دو فضای داخلی را ایجاد میکنند: ماتریکس داخلی و فضای بین دو غشا که بسیار باریک است. غشای داخلی چینخورده و تعداد زیادی کریستا ایجاد میکند که کل سطح آنرا بمقدار زیادی افزایش میدهد. سطح وسیع غشای داخلی، آنزیمهای دستگاه مولد انرژی میتوکندریایی (زنجیره تنفسی) را در خود جای داده است. ماتریکس میتوکندری واجد نسخههای یکسان متعددی از ژنوم میتوکندری، ریبوزومهای ویژه میتوکندری (میتوریبوزوم)، tRNAها و آنزیمهای متنوعی است که برای بیان ژنهای میتوکندری مورد نیازند. (۲۰)
ژنوم میتوکندری انسان:
حضور DNA در میتوکندری در سال ۱۹۶۳ و با استفاده از میکروسکوپ الکترونی مشخص شده است. DNA میتوکندریایی انسان یک ملکول مدور بسته دو رشتهای با ۱۶۵۶۹ جفت نوکلئوتید است. دو رشته mtDNA که به رشتههای H (سنگین) و L (سبک) معروفند، یک عدم تقارن غیر معمول در ترکیب بازهایشان دارند. زنجیره H غنی از پورین است در حالیکه زنجیره L غنی از پیریمیدین میباشد. سبک و سنگین به تحرک متفاوت رشتهها در گرادیانهای سزیم کلراید قلیایی اطلاق میشود. mtDNA انسان یکی از متراکمترین و فشردهترین بخشهای اطلاعات ژنتیکی است. در mtDNA، اینترون وجود ندارد و حتی بعضی از ژنهای آن همپوشانی دارند. DNA میتوکندریایی انسان واجد ژنهایی برای سیزده پروتئین (که همگی زیر واحدهای کمپلکسهای آنزیمی زنجیره تنفسی هستند)، ۲۲ tRNA و دو rRNA است. پلیپپتیدهایی که توسط mtDNA کد میشوند عبارتند از: هفت زیر واحد از ۴۲ زیر واحد تشکیلدهنده کمپلکس I که عبارتند از: ND5, ND4 , ND3, ND۲, ND1 وND6 یک زیرواحد از یازده زیر واحد تشکیل دهنده کمپلکس III که همان cyt b است؛ سه زیر واحد از ۱۳ زیر واحد کمپلکس IV که عبارتند از: COI (سیتوکروم c اکسیداز)، و COII ؛ دو زیر واحد از ۱۶ زیرواحد کمپلکس V که عبارتند از: ATPase6 و ATPase8. سایر زیرواحدهای پروتئینی کمپلکسهای زنجیره تنفسی و نیز دیگر پروتئینهای میتوکندری، توسط ژنوم هسته کد شده و سپس به میتوکندری منتقل میشوند (حدود ۱۰۰۰ پلیپپتید). هر میتوکندری واجد ۱۰-۲ کپی از DNA میتوکندری است. میتوکندریها از این نظر که تحت کنترل دو سیستم ژنتیکی DNA هسته و DNA میتوکندری هستند، در بین ارگانلهای سلولی منحصر بفردند. توالی نوکلئوتیدی mtDNA ، ۶ تا ۱۷ برابر سریعتر از توالیهای ژنی DNA هستهای باز میشوند؛ دلایل متعددی در این مورد ارائه شده است: میتوکندریها فاقد سیستمهای ترمیمی DNA موجود در هسته هستند که این امر موجب کارآیی کم میتوکندریها در ترمیم آسیب DNA میشود؛ هیستونها در میتوکندری وجود ندارند؛ میتوکندریها بیش از ۹۰% اکسیژنی را که به سلول وارد میشود، مصرف میکنند و بنابراین رادیکالهای آزاد اکسیژن ترجیحاً موجب آسیب DNA میتوکندری میشوند. میزان بالای جهش در mtDNA، موجب ایجاد RFLPهای متعدد، واریانتهای نوکلئوتیدی ناحیه کد کننده و ناحیه کنترل کننده، واریانتهای کنفورماسیونی و واریانتهای طولی میشود. واریانتهای پلیمورفیک با ریشه قومیتی و جغرافیایی نمونهها مرتبطند. این مساله احتمالاً به این دلیل است که جهشهای mtDNA در جریان پراکنده شدن اجداد مادری، هنگامی که زنان به بیرون از آفریقا و به اقلیمها و قارههای مختلف مهاجرت کردند، انباشته شدهاند. (۱۶، ۲۰ و ۳۴)
میتوکندریها نیمه خودمختار هستند:
از آنجائیکه میتوکندریها قادر به همانندسازی ژنوم خود بوده و سیستمهای همانندسازی، رونویسی و ترجمه مربوط به خود را دارا هستند؛ لذا آنها در داخل سیتوپلاسم سلول انسان مثل ارگانیسمهای نیمه مستقل عمل میکنند. (۲۰ و ۳۴)
میتوکندریها وراثت مادری دارند:
وراثت mtDNA متفاوت از وراثت هندسی ژنهای هستهای بوده و در انسان کاملاً مادری است. انتقال پدری mtDNA در مردان، حتی با استفاده از متود ICSI (تزریق داخل سیتوپلاسمی اسپرم)، نیز نشان داده نشده است. این مساله تا حدود زیادی ناشی از این حقیقت است که تخم پستاندار واجد حدود یکصد هزار میتوکندری و mtDNA است، در حالیکه اسپرم واجد حدود یکصد mtDNA است. mtDNAهای اسپرم در هنگام باروری به زایگوت داده میشود؛ که در پیوندهای بین گونهای خاص باقی میماند. با این حال در پیوندهای درون گونهای میتوکندریهای اسپرم بطور انتخابی حذف میشوند. این حذف با این کشف که میتوکندریهای اسپرم بایوبیکوئیتین نشاندار میشوند، مرتبط است. احتمالاً یوبیکوئیتین میتوکندریهای اسپرم را نشانهگذاری میکند تا هنگام ورود به اووسیت تجزیه شوند. (۱۷، ۲۴ و ۳۴)
هنزوپلاسمی و تفکیک رپلیکاتیو:
موقعی که یک جهش در mtDNA سلول رخ دهد، جمعیت مخلوطی از ملکولهای نرمال و موتانت در داخل سلول بوجود میآید که این پدیده را هتروپلاسمی میگویند. اینکه در موقع تقسیم سلول mtDNA موتانت به کدام سلول دختر منتقل شود، شانسی است. بنابراین درصد mtDNA جهش یافته در ردههای سلولی مختلف میتواند به طرف موتانت خالص یا نرمال خالص (هموپلاسمی) پیش رود. این فرایند به تفکیک رپلیکاتیو معروف است. مادران با mtDNA هتروپلاسمیک نسبتهای متفاوتی از mtDNA نرمال و جهش یافته را به فرزندان منتقل میکنند. در اغلب اختلالات میتوکندریایی ناشی از موتاسیونهای نقطهای و حذفی، مخلوطی از mtDNA وحشی و موتانت یافت میشود.در هیبریدهای سلولی سوماتیک بین رده های سلولهای انسانی ترانس فورمه، جهت تفکیک ظاهراً تصادفی است. با این حال اگر هیبریداسیون بین سلولهای هلا و فیبروبلاستهای دیپلوئید و یا بین سلولهای هلا و ردههای سلولی لنفوبلاستی باشد، mtDNAهای هلا ترجیحاً از دست میروند که علت این تفکیک رپلیکاتیو جهتدار، مشخص نیست. ممکن است این مساله از نظر کلینیکی مهم باشد چرا که گزارش شده است که در سلولهای واجد موتاسیون tRNA بیماریزای MELAS 3243 G ، mtDNAهای وحشی بطور انتخابی از بین میروند و mtDNAهای جهش یافته ترجیحاً باقی میمانند. به هر حال قوانین تعیین کننده این تفکیک جهتدار mtDNA و فاکتورهای موثر در آن هنوز شناخته نشدهاند. (۲۰، ۲۴، ۳۴ و ۵۰)
نوترکیبی mtDNA :
از سالها قبل شواهدی مبنی بر اینکه مخلوط شدن mtDNA در داخل سلولهای سوماتیک ممکن است نوترکیبی ایجاد کند، وجود داشته است. هر چند تعداد دفعات نوترکیبی در سلولهای کشت شده پستانداران کم است، اما نوترکیبیهای درون ملکولی ممکن است مکرراً رخ دهند . اگر یک mtDNA واجد حذف به سلول زایای موش ماده وارد شود، منجر به ایجاد استرینی از موش میشود که در آن فرزندان واجد mtDNAهای نرمال و واجد حذف هستند. ملکولهای دوپلیکیت نیز افزایش مییابند که به نظر میرسد از ترکیب ملکولهای نرمال و واجد حذف بوجود آمده باشند. با این حال هیچ مشاهدهای مبنی بر وجود نوترکیبی در بین ردههای مختلف mtDNA انسانی وجود ندارد. بنابراین احتمالاً در بین ردههای mtDNA انسانی، نوترکیبی رخ نمیدهد. شاید علت این مساله حذف میتوکندریها و mtDNAهای اسپرم توسط اووسیت از طریق تجزیه با واسطه یوبیکوئیتین باشد. در نتیجه ردههای مختلف mtDNA انسانی، از نظر فیزیکی جدانگه داشته میشوند و هرگز از نظر فیزیکی آنقدر با هم تماس ندارند که منجر به نو ترکیبی شود. (۲۰-۲۴)
کامل شدن (complementation) mtDNA :
به نظر میرسد که میتوکندریها و mtDNAهای داخل یک سلول در هم ادغام میشوند و این ادغام موجب میشود تا ژنومهای mtDNA همدیگر را به صورت ترانس تکمیل کنند. این پدیده اولین بار با ادغام دو سلول انسانی با همدیگر و تشکیل هیبرید، نشان داده شده است. هر چند مشاهدات متعددی ادغام داخل سلولی میتوکندریها و تکمیل شدن mtDNA را تایید میکنند، اما تحقیقات بیشتری جهت توصیف این پدیده نیاز است.(۲۰)
میزان بالای موتاسیون در mtDNA :
میزان موتاسیون mtDNA بسیار بیشتر از ژنهای هستهایست. مقایسه تنوع توالی ژنهای nDNA و mtDNA که در آنزیمهای یکسانی عمل میکنند، نشان میدهد که ژنهای mtDNA حدود ۱۷-۱۰ برابر سریعتر از ژنهای nDNA متحول میشوند. این سرعت بالای تغییر توالی ناشی از تجمع طیف وسیعی از پلیمورفیسمهای توالی mtDNA، در ردههای مختلف افراد مونث در جمعیت انسانی است. هر چند پلی مورفیسمهای mtDNA شایعند، اما آنها بایستی خنثی و بیاثر باشند تا بوسیله تغییر تدریجی ژنتیکی در جمعیت ایجاد شوند. با این حال موتاسیونها تصادفی هستند و با در نظر گرفتن اینکه اغلب نوکلئوتیدها در mtDNA عملکرد کد کننده دارند، لذا درصد بالایی از موتاسیونهای mtDNA بایستی مضر باشند. این موتاسیونها ممکن است تعویضهای بازی یا نوآرایی باشند که در رده زایای مونث یا در تکامل اولیه ناشی از توزیع سیستمیک یا بیماری اتفاق میافتد آنها ممکن است در طول زندگی در بافتهای بدن ایجاد شده و بصورت گروهی از موتاسیونهای هتروژن در بافتهای post-mitotic ، تجمع یابند. این موتاسیونها اساس ملکولی بالا بودن فراوانی بیماریهای mtDNA است که در کلینیک آنها را مشاهده میکنیم. (۲۰، ۲۴، ۳۴ و ۳۹)
تنوع پلی مورفیک mtDNA در جمعیتهای انسانی:
از آنجاییکه mtDNA کاملاً به صورت مادری به ارث میرسد بنابراین توالی mtDNA تنها بوسیله تجمع تعویضهای بازی در جریان پراکندگی ردههای مادری دچار تغییر و تحول میشود. این بدان معناست که تغییرات mtDNA بایستی با مبدا جغرافیایی افراد، مطابقت داشته باشد. این مساله اثبات شده است چرا که آفریقاییها، آسیاییها و اروپاییها هر کدام (HpaI mtDNA RSPs) HpaI mtDNARestriction sitepolymor phisms پلیمورفیسم mtDNA ناشی از برش توسط HpaI) مجزای مخصوص قارهای دارند. تحقیقات بیشتر بر روی RSPها، نشان دادهاند که تمامی انواع mtDNA به یک درخت با تنوع بسیار زیاد mtDNA متعلقند، که ریشه این درخت در آفریقاست و شاخههای آن به قارههای مختلف پراکنده شدهاند. تنوع توالی که در یک mtDNA خاص یافت میشود، هاپلوتیپ mtDNA و یک گروه از هاپلوتیپهای مرتبط با هم، هاپلوگروپ نامیده میشوند. هاپلوگروپها بوسیله پلی مورفیسمهای توالی قدیمی که ناشی از پراکندگی یک شاخه خاص از درخت mtDNA است تعریف میشوند. ساختار و پراکندگی درخت mtDNA در دو مطالعه توالیهای کامل mtDNA که مبدا آفریقایی mtDNA و شاخههای قارهای به ترتیب از آفریقا به آسیا و به اروپا را به وجود آورده و مشخص کرده است، ایجاد شده است.
فهرست مطالب
مقدمه
ساختار میتوکندری
ژنوم میتوکندری انسان
میتوکندریها نیمه خود مختار هستند
میتوکندریها وراثت مادری دارند
هتروپلاسمی و تفکیک رپلیکاتیو
نوترکیبی mtDNA
کامل شدن mtDNA
میزان بالای موتاسیون در mtDNA
تنوع پلی مورفیک mtDNA در جمعیتهای انسانی
ژنتیک میتوکندری (همانندسازی، رونویسی و ترجمه mtDNA)
فرایندهای میتوکندریایی
میتوکندری و پاسخ به استرس
بیان آستانهای
بیماریهای میتوکندریایی ناشی از جهشهای سیستمیک
LHON (نوروپاتی چشمی ارثی لبر)
مکانیسمهای پاتوفیزیولوژیکی احتمالی LHON
LHON، مولتیپل اسکلروزیس و دیستونی
بیماری پارکینسون (PD) و بیماری هانتینگتون (HD)
ژنتیک کروموزومی بیماری پارکینسون
جهشهای mtDNA در PD
اختلالات میتوکندریایی در PD
نارساییهای میتوکندریایی در بیماری هانتینگتون
رتینیت پیگمنتوزا (RP) و سندرم لی (LS)
موتاسیونهای mtDNA در RP و سندرم لی
میوپاتی و انسفالومیوپاتیهای میتوکندریایی
ضعف عضلانی پیشرونده و مرتبط پاموتاسیونهای سیتوکروم mtDNA b
انسفالومیوپاتیهای ناشی از جهشهای ژن COX mtDNA
میوپاتیهای میتوکندریایی ناشی از موناسیونهای TRNA ژنوم میتوکندری
کاردیو میوپاتیهایپرتروفیک و میوپاتی ناشی از جهشهای mtDNA
انسفالومیوپاتیهای ناشی از جهشهای mtDNA
افتالموپلژیا، پتوزیس و میوپاتی میتوکندریایی
افتالموپلژیای ناشی از جهشهای mtDNA
CPEO و KSS مرتبط با موتاسیونهای نوآرایی mtDNA
CPEO ناشی از موتاسیونهای تعویض باز mtDNA
سندرم مغز استخوانی پانکراسی پیرسون
دیابت ملیتوس
تیپ II دیابت ملیتوس بوسیله نوآراییهای (حذفها و دوپلیکاسیونها) mtDNA
ایجاد میشود
دیابت تیپ II ناشی از موتاسیونهای تعویض باز mtDNA
میوپاتی و دیابت
پاتوفیزیولوژی دیابت و کری
کری به ارث رسیده از مادر و یا کری القا شده توسط آمینوگلیکوزید
دمانس بعنوان یک بیماری میتوکندریایی
بیولوژی و ژنتیک بیماری آلزایمر
اختلالات میتوکندریایی در AD
بیماری آلزایمر ناشی از جهشهای mtDNA
دیس کندروپلاژی متافیزی یا هیپوپلازی مویی- غضروفی ناشی از جهشهای
RNASE MRP
بیماریهای مولتی فاکتوریال و mtDNA
جهشهای سوماتیک mtDNA در بیماریهای دژنراتیو، سرطان و پیری
تجمع جهشهای سوماتیک mtDNA مرتبط با سن
آنمی سیدروبلاستیک ایدیوپاتیک
بیماری ایسکمی قلبی و کاردیومیوپاتی اتساعی
بیماریهای نورودژنراتیو؛ HD, PD و AD
بیماری پارکینسون و بیماری هانتینگتون
بیماری آلزایمر
موتاسیونهای سوماتیک mtDNA در دیگر بیماریهای کمپلکس
موتاسیونهای سوماتیک در سرطان
نتیجهگیری
منابع
منابع:
۱- Allen J.C., Midhailovskay I.E., Sukernik R.I., Wallace D.C.; The role of mtDNA background in disease expression: a new primary LHON mutation associated with western eurasian haplogroup J. Hum Genet; 2002; 110(2): 130-138.
۲- Alcolado J.C., thomas A.W.; Maternally inherited diabetes mellitus: the role of mitochondrial DNA defects. Diabet Med; 1995;12:102-108.
۳- Brown M.D., Voljavec A.S., latt M.T., Wallace D.C.; Leber’s hereditary optic neuropathy: A model for mitochondrial neurodegenerative disease. FASEB.J; 1992b; 6: 2791-2799.
۴- Bianca J.C., Rene F.M., Jeroen G., Marianne d.,Hubert J.;Mutation analysis of the entire mitochondrial genome using denaturing high performance liquid chromatography. Nucleic Acids Research; 2000; Vol. 28; No.20 : 89.
۵- Bruno C.,Martinuzzi A., Tang y.;A stop-codon mutation in the human mtDNA cytochromeC oxidaseI gene disrupts the functional struture of complex IV. Am.J. Hum. Genet.; 1999; 65:611-620.
۶- Brown M.D., Wallace D.C.; Molecular basis of mtDNA disease. J.Bioenerg. Biomembr; 1994a; 26: 273-289.
۷- Brown M.D., Shoffner J.M., Kim Y.L.; Mitochondrial DNA sequeca analysis of four Alzheimer’s and parkinson’s disease patients. Am.J. Hum. Genet; 1996; 61: 283-289.
۸- Bindoff L.A., Brich M.,parker W.D., Tunbull D.M.; Mitochondrial function in parkinson’s disease. Lancet; 1989;2:49.
۹- Brockington M.,Alsanjari N.,Sweeny M.G.,Harding A.E.; kearns-sayre syndrome associated with mitochondrial DNA deletion or duplication:A molecular genetic and pathological study. J.Neurol. sci; 1995; 131: 78-87.
۱۰-Brennan W.A., Bird E.D., Aprille J.R.; Regional mitochondrial respiratory activity in Huntington’s disease brain. J.Neurochem.; 1985;44: 1948-1950.
۱۱-Bindoff L.R., Howell N.,Poulton J.; Abnormal RNA processing associated With a novel tRNA mutation in mitochondrial DNA:A potential disease mechanism. J.Biol. chem.; 1993;268 : 19559-564.
۱۲-Blin O.,Desnuelle C., Rascol O.; Mitochondrial respiratory failure in skeletal muscle from patients with parkinson’s disease and multiple system atrophy. J. Neurol. Sci.; 1994; 125:95-101.
۱۳-Bofol D., Scacco S.C., Solarino G., Papa s.; Decline with age of the respiratory chain activity in human skeletal muscle. Biochim.Biophys. Acta.; 1994; 1226:73-82.
۱۴-Bonilla E., Tanji K., Hirano M., Vu t.H., Dimauro s., schon E.A.; Mitocondrial involvement in Alzheimer’s disease. Biochim. Biophys. Acta; 1999; 1410:171-182.
۱۵-Boulet L., karpati G., shoubridge E.A.; Distribution an thre shold expression of the tRNALys mutation in skeletal muscle of patients with myoclonic epilepsy and ragged- red fibers (MERRF). Am.j.Hum. Genet.; 1992; 51 :1187-1200.
۱۶-Cann R.L., stoneking M., Wilson A.c.; Mitochondrial DNA and Human evolution. Nature; 1987;325:31-36.
۱۷-Case J.T.,Wallace D.C.; Maternal inheritance of mitochondrial DNA polymorphisms in cultured human fibroblasts. Somatic Cell Genetic; 1981;7:103-108.
۱۸-Carelli V.,Ghelli A.,RattaM.; Leber’s hereditary optic neuropathy: Biochemical effect of 11778/ND4 and 3460/ND1 mutations and correlation with the mitochondrialgenotype. Neurology; 1997; 48:1623-1632.
۱۹-Chinnery P.F., Turnbull D.M.; Mitochondrial DNA disease. Lancet; 1999 ; 354: 17-21.
۲۰-David L.R imoin, Michael C.,Reed E.; EMERY and RIMOIN’S principles and practice of Medical Genetics. Fourth edition;2001.
۲۱-Dumoulin R.,Lopez R.F., Barker W.W.; Anovel gly 290 asp mitochondrial cytb mutation linked to a complex III deficieny in proressive exercise intolerance. Molecular and cellular probes; 1996 ; 10:389-391.
۲۲-Enriguez J.E., chomy A., and Attardi G.; MtDNA mutation in MERRF syndrome causes defective aminoacylation of tRNALys and premature translation termination. Nature. Genet.;1995; 10:
۴۷-۵۵٫
۲۳-Francesca O., Michele R., Anna M., Pietro T., Gabriella S.; Pathogenic role of mtDNA duplications in mitochondrial disease associated with mtDNA deletions. American Journal of Medical Genetics; 2003;118(3): 247-254.
۲۴-Houshmand M.;Mitochondrial DNA mutations, pathogenicity and inheritance. Iranian Journal of Biotechnology; 2003; vol.1; No.1.
۲۵-Herman A.C., Benttage and Giuseppe Attaydi; Relationship of genotype to phenotype in fibroblast-derived transmitochondrial cell lines carrying the 3243 mutation associated with the MELAS encephalomyopathy: shift towords mutamt genotype and role of mtDNA copy number. Human Molecular Genetics; 1996 ; 5(2):197-205.
۲۶-Josep L.; Pathology of the mitochondrion. Autonomous university of Barcelona, Barcelona, spain; 2003.
۲۷-Johns D.R.,Neufeld M.J., porrk R.D.; An ND mitochondrial DNA mutation associated with leber hereditary optic neuropathy. Biochem. Biophys. Res. Commun.; 1992a; 187:1551-1557.
۲۸-Kadowaki T., Sakura H., Otabe S.; Asubtype of diabetes mellitus associated with a mutation in the mitochondrial gene. Muscle and Nerve; 1995;3:137-141.
۲۹-Kanamori A., Tanaka K., Umezava S.; Insuline resistance in mitochondrial gene mutation. Diabetes care; 1994; 17: 778-779.
۳۰-King M.P., Koga Y.,Davidson M.,Schon E.A.; Defects in mitochondrial protein syntesis and respiratory chain activity segregate with the tRNALeu(uuR) mutation associatod with mitochondrial myopathy, encephalopathy, Lactic acidosis, and strock like episodes. Mol. Cell. Biol.; 1992; 12:480-490.
۳۱- Lu C.Y., Lee H.C.,Fahn H.J., Wei Y.H.; Oxidative damage elicited by imbalance of free redical scavenging. Enzymes is associated with large- scale mtDNA deletions in aging human skin- skin. Mutat,Res.; 1999; 25(2):11-21.
۳۲-Lenaz G.; Role of mitochondria in oxidative stress and aging Biochim. Biophys. Acta.; 1998; 1366: 53-67.
۳۳-Lee H.C., wei Y.H.; Mutation and oxidative damage of mtDNA and defective turnover of mitochondria in human aging. J.Formos. Med.Assoc.; 1997; 96:770-800.
۳۴-Lois A.,Tully C., Barbara C., Levin; Human mitochondrial Genetics. Biotechnology and Genetic Engineering Reviews; 2000; Vol.17.
۳۵-Lee H.C., Wei Y.H.; Mitochondrial alterations, cellular response to oxidative stress and defective degradation ot proteins in aging. Biogerontology; 2001; 2(4): 231-240.
۳۶-Myabayashi S.,Nakamura R., Tada K.; Defects of mitochondrial respiratory enzymesincloned cells from MELAS fibroblasts. J. Inher. Metab. Dis.; 1992;15:797-802.
۳۷-MtDNA in aging, cancer and mitochondrial disease. Innovita Research Fundation; 2003.
۳۸-MtDNA change during aging. Innovita ,Research Fundation; 2003.
۳۹-Rew D.A.; Mitochondrial DNA , human evolution and the cancer genotypo. EJSO; 2001; 27:209-211.
۴۰-Rew D-A.; Tahe evolutionary biology of aging, death and human malignancy. Eur.J.Surg. Oncol.; 1998;24:568-72.
۴۱-Richardson A.; Increased oxidative damage is correlated to alterd mitochondrial function in heterozygous manganese superoxide dismutase knockout mice. J.Biol. chem.; 1998; 237: 28510-28515.
۴۲-Schon E.A., Hirano M., Dimauro S.; Mitochondrial encepha lomyopathies: clinical and Molecular analysis. J.Bioenerg. Biomem.; 1994; 26: 291-29.
۴۳-Sastre J., pallardo F.V., Vina J.; Mitochondrial oxidative stress plays a key role in aging and opoptosis. IUBMB Life; 2000; 49(5): 427-35.
۴۴-Sastre J.,Pallardo F.V., Garcia d.,Vina J.; Mitochondria, oxidative stress and aging. Free, Radic. Res; 2000; 32(3): 189-198.
۴۵-Wei Y.H., Lee H.C.; oxidative stress, mitochondrial DNA mutation and Impairment of antioxidant enzumes in aging. Exp. Biol. Med.; 2002; 227(9): 671-820.
۴۶-Wei Y.H., Ma Y.S., Lee H.C., Lu C.Y.; Mitochondrial theory of aging matures- roles of mtDNA mutation and oxidative stress in human aging. Zhonghua. Yi. Xue. Za. Zhi (taipei); 2001; 64(5):259-70.
۴۷-WallacD.C.; Mitochondrial DNA mutations in diseases of energy metabolism. J.Bioenery. Biomem; 1994;26 : 241-250.
۴۸-Wei Y.H.; oxidative stress and mitochondrial DNA mutations in human aging. Proc. Soc. Exp.Biol. Med.; 1998; 217(1):53-63.
۴۹-Wallace D.C.;Mitochondrial DNA sequence Variation in human evolution and disease. Proc. Natl. Acad. Sci. USA.; 1994;91:8739-8746.
۵۰-White F.A., Bunn C.L.; Segregation of mtDNA in Human somatic cell by hybrides. Mol. Gen. Genet.; 1984; 197; 453-46.
۵۱-Yoshino H.,Nakagave Y., Kondo T.,mizuno Y.; Mitochondrial complex I and II activities of lymphocytes and platelets in parkinson’s disease. J. Neur., Trans. Parrkinsons. Dis. Dement. Sect.; 1992; 2: 27-34.
۵۲-Zeviani M., Muntono F., Savarese N.;A MERRF MELAS over lap syndrome associated with a new point mutation in the mitochondrial DNA tRNA Lys gene. Eur.J.Hum. Genet.; 1993; 1: 80-87.
۵۳-Zhang C., Baumer A., Maxwell R.J., Nagley p.; Multiple mtDNA deletions in an elderly human individual. FEBS; 1992:4-8.
![]()
![]() فرمت فایل: WORD
فرمت فایل: WORD
![]()
![]() تعداد صفحات: 132
تعداد صفحات: 132
مطالب مرتبط